Figure 1: The user interface of CAR.
CAR (short for "Contig Assembly using Rearrangements") is an efficient and more accurate tool for assembling contigs of a prokaryotic draft genome based on a reference genome. Given a set of contigs in multi-FASTA format and a reference genome in FASTA format, CAR can output a list of scaffolds that are groups of contigs whose relative placement and orientation are determined.
CAR provides a user interface (refer to Figure 1) that is intuitive and easy to operate. It takes as input a set of contigs of a draft chromosome in multi-FASTA format and a reference chromosome in FASTA format. For convenience, the user can choose one of the examples (1) we prepared in advance for running CAR, or submit a job according to the procedures described as follows.
Figure 1: The user interface of CAR.
In the output page (see Figure 2 for an example), CAR first shows the input data and user-specified parameter (8), including the nucleotide sequences of input draft and reference chromosomes, a dot plot graph between them before performing contig assembly (9), and the user-specified parameter of identifying conserved genetic markers. In the dot-plot graph (see Figure 3 for an example), the contigs of draft chromosome are plotted on the y-axis, while the sequence of reference chromosome is plotted on the x-axis. Moreover, the forward matches are displayed in red, while the reverse matches are displayed in blue. Next, CAR shows a contig assembly result of draft chromosome based on reference chromosome (10), including total running time (11), a set of scaffolds (12) (click here for an example) and its corresponding multi-FASTA file (13), a dot plot graph between assembled draft chromosome and reference chromosome (14) (see Figure 4 for an example), and a comparison of dot-plot graphs between before contig assembly and after contig assembly (15).

Figure 2: A display of output page of CAR
when running with a draft chromosome with nucleotide sequences.
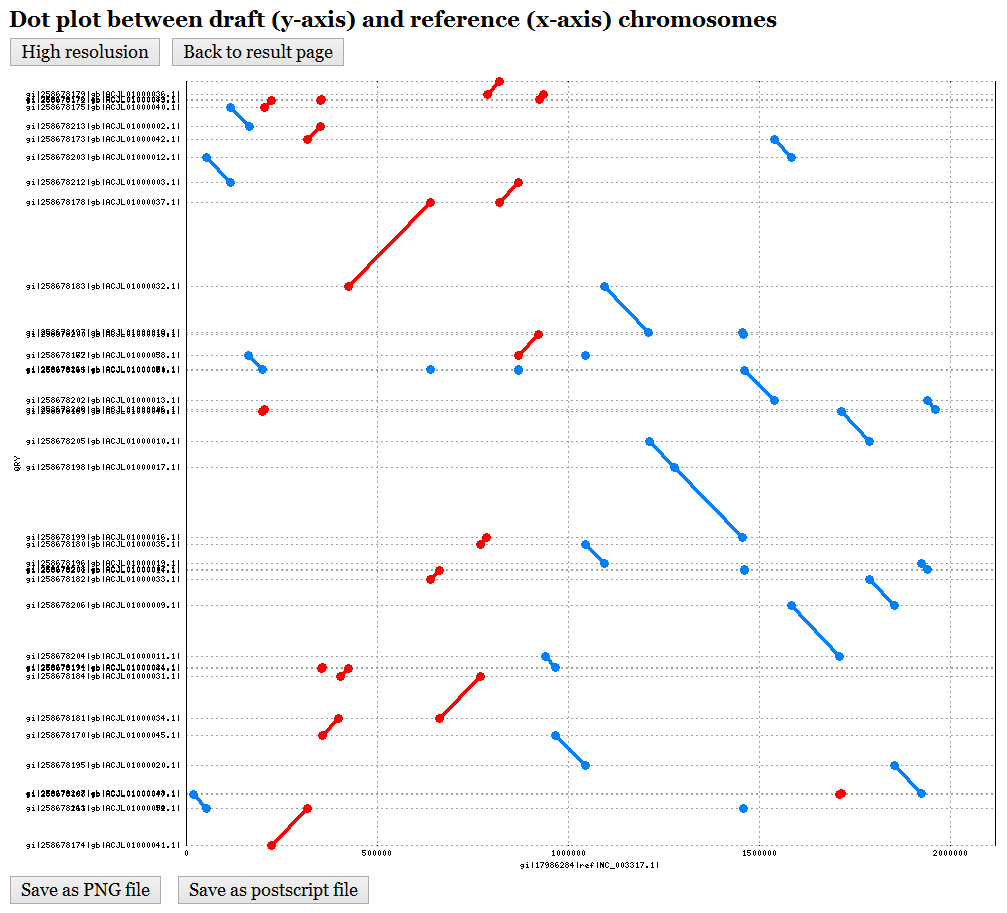
Figure 3: The dot plot of draft
and reference chromosomes before contig assembly.
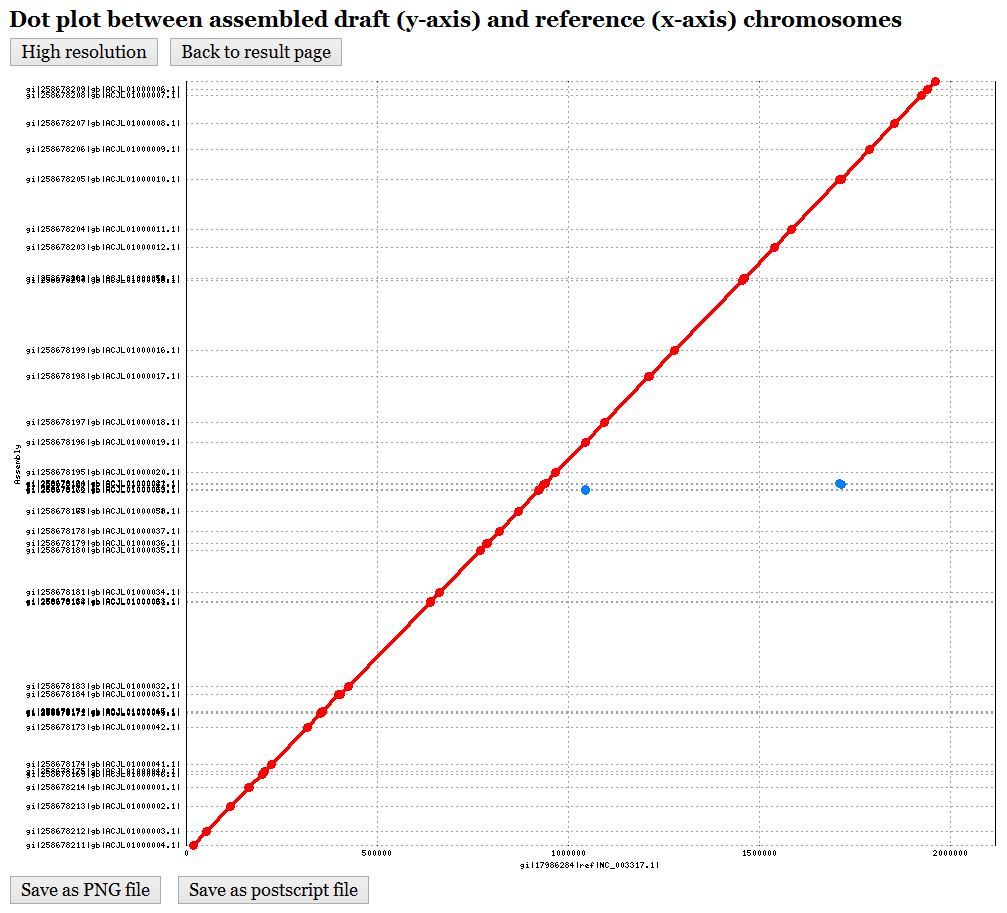
Figure 4: The dot plot of assembled draft
and reference chromosomes after contig assembly.
In the following table, we provide the results of CAR when running on some prokaryotic draft chromosomes. CAR can finish its assembly job in several seconds or a couple of minutes.
| CAR result of example 1 (15.14 sec) | CAR result of example 2 (16.91 sec) |
| CAR result of example 3 (14.96 sec) | CAR result of example 4 (16.28 sec) |
| CAR result of example 5 (30.94 sec) | CAR result of example 6 (93.55 sec) |
To validate CAR, we used a real dataset (click here for download) composed of several prokaryotic genomes to test it and compared its accuracy performance to eight other reference-based tools of contig assembly, namely Projector2 (van Hijum et al., 2005), OSLay (Richer et al., 2007), ABACAS (Assefa et al., 2009), Mauve Aligner (Rissman et al., 2009), fillScaffolds (Munozet al., 2010), r2cat (Husemann et al., 2010), CONTIGuator (Galardini et al., 2011) and SIS (Dias et al., 2012). This real dataset was used in the study of SIS by Dias et al., 2012, which contains 19 draft genomes of phylogenetically diverse prokaryotes that can be downloaded from the GenBank of NCBI. Among these 19 prokaryotic genomes, four of them have two chromosomes, while the others have only one, thus creating a total of 23 chromosomes in this dataset. Each of these 23 chromosomes was processed separately by each contig assembly tool. For the draft of each query chromosome, we used other 20 closest genomes (excluding the query chromosomal genome itself), which were also selected by Dias et al. (2012) from complete prokaryotic genomes at the GenBank of NCBI according to their phylogenetic distances from the query chromosomal genome, to serve as different reference genomes. The purpose for choosing 20 closest other genomes instead of only the closest is to understand how the accuracy performance of a contig assembly tool changes with increasing evolutionary distances between query and possible reference genomes.
In the studies of genome rearrangements (Fertin et al., 2009), a genetic marker (i.e., DNA sequence, gene or syntenic segment) usually is represented by a signed integer, in which the sign indicates the transcriptional direction (or strandedness) of the corresponding genetic marker.
Reversals, also called inversions, affect a block of consecutive genetic markers on a chromosome by reversing the order and flipping the signs of the corresponding integers. For example, the reversal depicted in Figure 5 rearranges a chromosomal segment of three genetic markers (1, -3) into (3, -1).
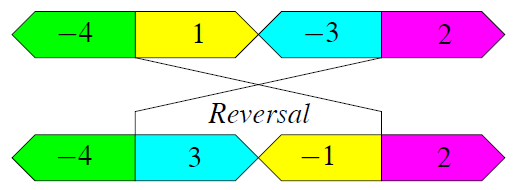
Figure 5: A reversal rearranges the chromosomal segment
(1, -3) into (3, -1).
Transpositions affect a chromosome by moving a block of its consecutive genetic markers to another location, as illustrated in Figure 6 for an example, or equivalently by exchanging two adjacent and non-overlapping blocks of consecutive genetic markers on the chromosome.
![]()
Figure 6: A transposition moves the block of a single genetic marker 1
into a new location between genetic markers -3 and 2 or, equivalently,
exchanges two adjacent and non-overlapping block (1) and (-3).
This is a special case of block-interchange.
Block-interchanges are a kind of generalized transpositions that exchange two non-overlapping but not necessarily adjacent blocks of consecutive genetic markers on a chromosome. Therefore, the rearrangement exemplified in Figure 6 are both a transposition and a block-interchange, but the one shown in Figure 7 is just a block-interchange, but not a transposition.

Figure 7: A block-interchange (also called generalized transposition)
exchanges two non-adjacent blocks (-4) and (2).
Notice that this is not a transposition.
A sequence in FASTA format starts with a single-line description,
followed by lines of sequence data. The description line starts with
a right angle bracket (">") and is usually followed by the sequence
identifiers and description.
An example of a FASTA-format sequence is given as follows.
>sequence TTGACCGATGACCCCGGTTCAGGCTTCACCACAGTGTGGAACGCGGTCGTCTCCGAACTTAACGGCGACC CTAAGGTTGACGACGGACCCAGCAGTGATGCTAATCTCAGCGCTCCGCTGACCCCTCAGCAAAGGGCTTGA multi-FASTA format consists of multiple FASTA-format sequences. The following is an example.
>sequence1 CAAAACGAAATCGAGCGCCATCTGCGGGCCCCGATTACCGACGCTCTCAGCCGCCGACTCGGACATCAGA >sequence2 TCCAACTCGGGGTCCGCATCGCTCCGCCGGCGACCGACGAAGCCGACGACACTACCGTGCCGCCTTCCGA >sequence3 AAATCCTGCTACCACATCGCCAGACACCACAACCGACAACGACGAGATTGATGACAGCGCTGCGGCACGG